Introduction: The rise of immune-modulating therapeutics
Modern pipelines rely more and more on modalities that influence the immune system in complex ways. Therapeutic proteins with tailored Fc domains, PEGylated constructs, oligonucleotides, liposomal and micellar formulations, and other engineered biologics are designed to correct dysregulated immunity or to harness it for therapy. The same features that make these compounds powerful can also provoke unintended immune activation in first-in-human studies.
Adverse immunostimulation in early human trials
Clinical teams recognize this as adverse immunostimulation (AIS), a broad label for acute, often unexpected activation of immune pathways that produces symptoms ranging from flushing and urticaria to hypotension, fever, chest discomfort, and laboratory shifts such as cytokine spikes or complement consumption. Prediction from preclinical packages still remains imperfect, in part because species sensitivity differs and in part because common bench assays only approximate the complex biology that unfolds in human blood and tissue.
Yet, if early clinical work is deliberately instrumented to learn, using harmonized in vivo challenges, ex vivo whole-blood and serum analyses, and fit-for-purpose in vitro assays, investigators can both protect participants and extract mechanism-level knowledge that keeps programs moving.
A structured framework from CHDR
Centre for Human Drug Research (CHDR) set out the case for this approach and shows how to make it operational. We summarize the experience from three first-in-human programs in which AIS occurred despite careful adherence to guidance and thoughtful preclinical work. We also translate those lessons into a practical, clinic-ready framework for recognition, sampling, interpretation, and mitigation. What matters most is not a promise that AIS will not happen, but a shared plan that, if it does, the study will capture the right data at the right times and will have clear rules for safe continuation or re-exposure.
Distinguishing mechanisms beneath similar clinical signs
The main issue is that the bedside picture of AIS can look similar across mechanisms, yet the biology underneath is different. For instance:
IgE-mediated reactions involve sensitization, mast cell and basophil degranulation, and a characteristic rise in tryptase, sometimes with histamine transients and supportive evidence from specific IgE.
Complement activation-related pseudo-allergy (CARPA) depends on activation of the complement cascade with the generation of anaphylatoxins such as C3a and C5a, leading to mast cell and macrophage activation without prior sensitization.
Cytokine release syndrome is driven by the acute production of mediators such as IL-6, TNF, and IFN-γ by monocytes, macrophages, and other Fc receptor bearing cells, and can progress to capillary leak physiology.
At the bedside these pathways can blur, which is why standardized sampling windows and analyte panels are so important.
Sampling strategies and operational details
The CHDR framework recommends collecting baseline samples when a priori risk is present, sampling at the onset of symptoms, and sampling again at resolution, with attention to preanalytical handling. Complement activation products degrade quickly, so plasma needs to be kept cold and processed without delay. Tryptase peaks early and declines within hours, so timing matters if an IgE mechanism is suspected. Cytokines can rise fast, whereas CRP tends to lag. These small operational details determine whether the data will resolve the mechanism or leave teams guessing.
Proactive planning before first dosing
Planning should begin before the first dosing. Sponsors and investigators should sit together and describe, in plain language, where the risk might come from in this specific program. Is the construct likely to engage complement because of its surface chemistry or aggregation profile, is Fcγ engagement central to the mode of action, or are there sequence motifs that might trigger pattern recognition receptors on innate cells?
That discussion should end in concrete study procedures: which tubes to draw and when, where those samples will go, and what thresholds will trigger predefined actions such as pausing an infusion, stepping down the rate, or initiating premedication for re-exposure. It should also cover documentation of skin findings and orthostatic vitals, observation windows to detect biphasic reactions, and the practicalities of keeping IV access in place and the participant supine during acute management. By aligning on these details early, teams remove the uncertainty that often leads to conservative holds and long interruptions when symptoms first appear.
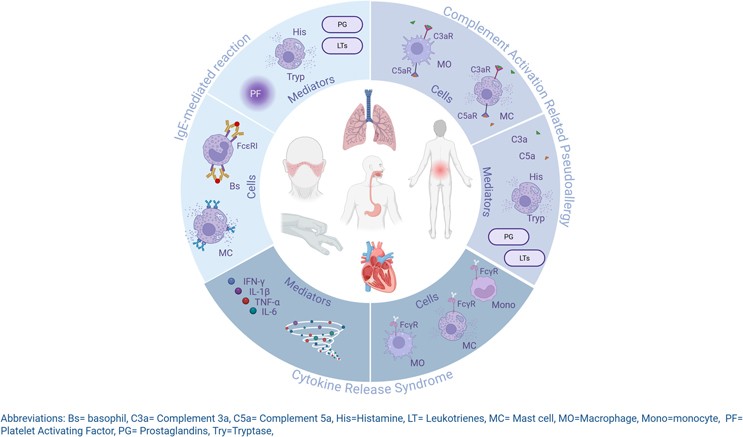 Image created with BioRender.com
Image created with BioRender.com
In vivo immune challenges as tools for learning
Once the clinic is equipped to learn, in vivo immune challenges become powerful tools to reveal both intended pharmacology and unintended responses under controlled conditions. Intravenous or intradermal endotoxin activates innate immunity in a predictable, short-lived manner, providing a clear background to assess anti-inflammatory effects. Keyhole Limpet Hemocyanin (KLH) elicits a de novo adaptive response that can be tracked systemically and locally, while topical imiquimod or UVB exposure engages skin-resident immunity as a repeatable tissue model. Each challenge is chosen to fit the drug’s mechanism and sampling feasibility, and, crucially, is integrated with AIS monitoring so that the same standardized sampling clarifies both adverse reactions and desired pharmacodynamic effects.
Ex vivo approaches to bridge bench and clinic
The ex vivo layer bridges clinic and bench. Whole-blood assays preserve physiological context and help identify the mechanism behind immune reactions. Elevated C3a and C4a with normal tryptase indicate complement activation rather than IgE involvement, while rises in IL-6 and TNF point to cytokine release. Using each volunteer’s baseline enables precise interpretation and supports safe re-exposure. Such clarity is only achievable when ex vivo sampling is built into the protocol from the outset.
In vitro assays to anticipate liability
Fit-for-purpose in vitro assays complete the assessment by identifying compound-specific immune liabilities before dosing. Cytokine release panels, Fcγ receptor binding tests, complement activation screens, and TLR reporter assays each address distinct mechanisms and should be tailored to the compound’s structure, target, and formulation. Including clear controls and predefined readouts ensures meaningful results. Insights from these assays guide practical clinical decisions, such as infusion speed, dose steps, or premedication with antihistamines or antipyretics, so that immune activation becomes a measurable and manageable event.
Case experiences demonstrating the framework
Real study experience from CHDR shows why this structure matters.
Modified Fc monoclonal antibody: This candidate looked benign in animal studies but produced infusion rate-dependent urticaria and orthostatic hypotension within two hours at a higher human dose. Complement activation products rose in vivo, while tryptase and IgE did not, and cytokines remained unremarkable. Slowing the infusion and adding premedication reduced symptoms but did not eliminate them.
Oligonucleotide mixture: Despite clean toxicology, this compound produced fever and headache at the top dose in a small subset of study participants, accompanied by mild, transient leukocytosis and modest elevations in cytokines.
Intact Fc monoclonal antibody: This molecule produced a clearer cytokine pattern at higher cohorts, with fever, chest discomfort, laboratory increases in IL-6, TNF, and IL-8, and improvement when the infusion rate was reduced and premedication added.
None of these reactions was predicted with certainty by preclinical work. All became manageable because sampling, handling, and decision rules were already in place, and because the clinical team knew what biomarkers would separate one mechanism from another.
Regulatory context and collaborative practice
Regulatory guidance recognizes that standard animal toxicology often falls short for biotechnology products and calls for mechanism-based immunotoxicity assessments. Clinical playbooks turn these principles into concrete practice, which should define sampling methods, timing, and clear decision criteria while promoting shared responsibility between sponsor and investigator. This collaboration prevents minor immune reactions from causing unnecessary study delays. By planning ahead, teams can ensure safety without losing momentum, gain early proof of pharmacology, and distinguish between IgE, complement, or cytokine-driven responses to guide dosing and mitigation strategies.
Advanced tools for richer immunological profiling
CHDR’s immunology and inflammation toolset illustrates how to implement this without overcomplicating studies. Endotoxin, KLH, imiquimod, and UVB challenges are all established, safe in experienced hands, and highly informative when paired with modern analytics. Blood and skin can be sampled at several time points, and transcriptomic, proteomic, and spatial pathology methods provide deep readouts that map onto clinical signs and standard safety labs. Ex vivo and in vitro assays are planned with the same care as pharmacokinetics, so the results arrive when clinical teams need to make decisions rather than months later.
Conclusion: toward smarter early development
Early immune profiling is about asking the right questions in the right systems and learning from every response. At CHDR, this approach combines in vivo challenge models with ex vivo and in vitro analyses to reveal how new therapies interact with human immunity in real time. Together, this allows study teams to anticipate risk, to refine dosing, and understand pharmacology from the very first study in humans. By including this strategy into early development, CHDR helps sponsors move forward with confidence, building evidence-based clinical development programs.
References
van den Noort JA, van Ruissen MCE, Smidt LCA, Klarenbeek NB, Kremer PHK, Jansen MAA, Burggraaf J, van Gerven JMA, Moerland M. Adverse immunostimulation in early phase clinical trials: Key findings and recommendations based on the investigator’s clinical experience. British Journal of Clinical Pharmacology. First published 19 September 2025. doi:10.1002/bcp.70276.
van den Noort JA, Meijs AC, Klarenbeek NB, Moerland M. Navigating adverse immunostimulation: A practical guide for clinical researchers. British Journal of Clinical Pharmacology. First published 12 September 2025. doi:10.1002/bcp.70268.
